Placing the molecular properties of cancer in the context of the patient
Most cancer research has focused on the molecular properties of the tumor and its microenvironment in order to therapeutically target the components that drive disease progression. But cancer cells do not exist in isolation. For roughly 20% of breast cancer patients, the tumor and its microenvironment do not carry enough signal to predict how the disease will progress, and no tumor-site marker alone reliably tracks response to standard-of-care treatment or long-term prognosis. Other factors must be playing a role: the patient’s immune system, the gut microbiome, lifestyle, and environmental exposures.
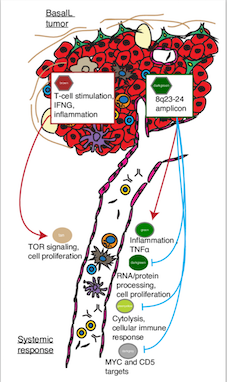
In our most recent study, we identified molecular processes in the primary breast tumor that are tightly co-expressed with processes in the patient’s blood cells — our window onto their systemic immune response. RNA profiling of blood is a practical way to read the state of the immune system in an individual patient, and feeds directly into precision-medicine workflows.
To date, most of our work has relied on RNA harvested from whole blood, a mixture of many cell types including T and B lymphocytes, neutrophils, and platelets. The molecular profiles we get from these samples are therefore averages, blending the transcriptional programs of every cell type. Single-cell RNA sequencing now lets us read each cell type separately, which matters when tracking how an individual patient responds to therapy, develops adverse effects, or progresses toward recurrence.
The gut microbiome shapes a surprisingly wide range of host physiological processes, from metabolism to immune regulation. In parallel with our work on the immune system, we’ve started to characterize these complex microbial communities, asking how they are shaped by lifestyle and how they, in turn, might influence the host, for example by modulating the systemic immune response to cancer.
Taken together with information about a patient’s exposures and lifestyle, these interactions are a first step toward a new generation of integrative, holistic predictors that can support a broad range of clinical questions, including diagnosis, response to therapy, prognosis, and disease monitoring.
Precision oncology for in situ breast cancer (DCIS)
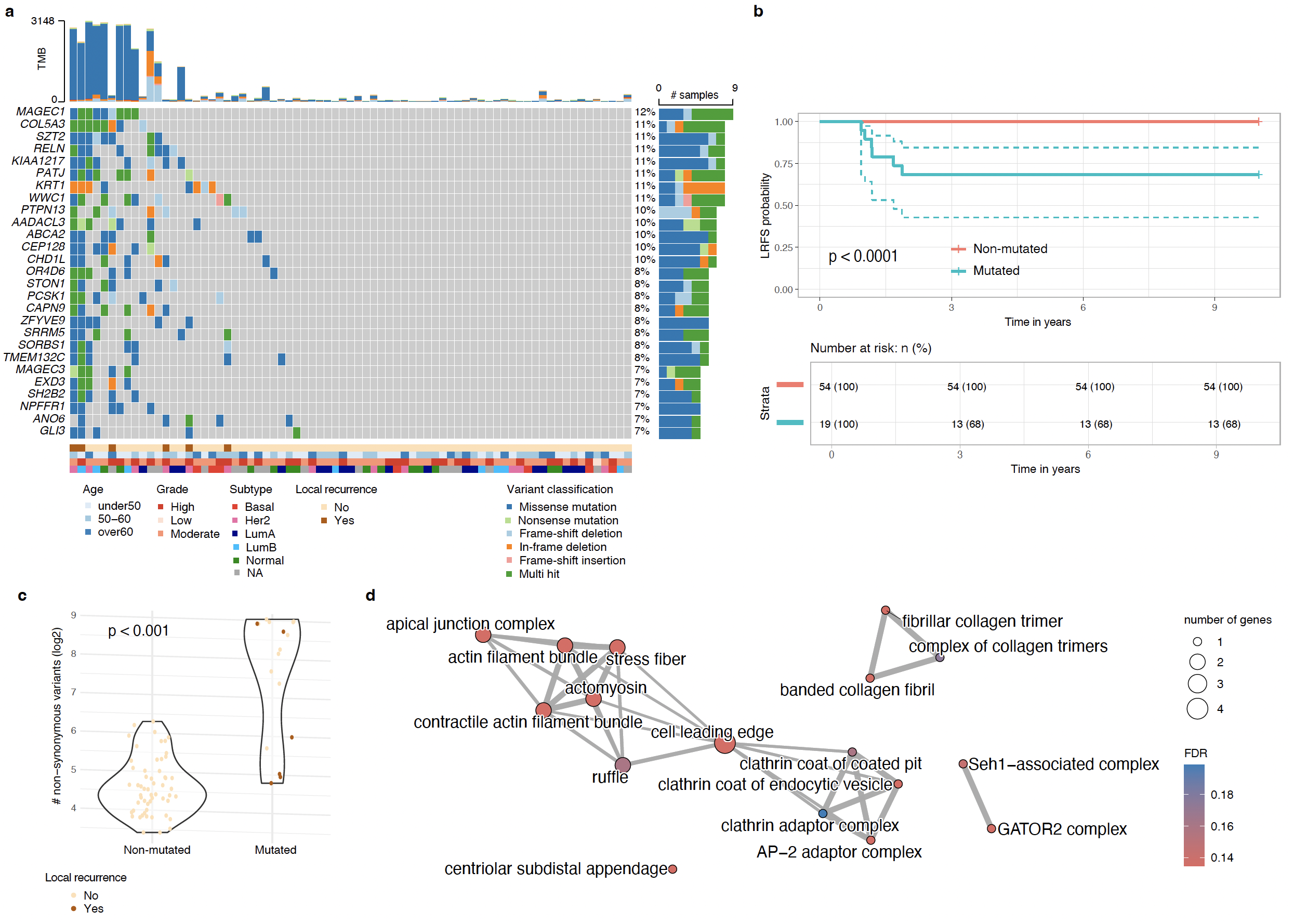
Ductal carcinoma in situ (DCIS) is a non-obligate precursor to invasive breast cancer. Most patients are treated with surgery and adjuvant radiotherapy (RT), yet the absolute benefit of RT for an individual patient is highly variable: some are over-treated for disease that would never have recurred, others develop invasive recurrence despite standard care. Better molecular risk stratification is needed both to de-escalate therapy in low-risk disease and to identify RT-resistant lesions that would benefit from alternative management.
Through whole-exome sequencing of pure DCIS FFPE specimens, we are characterizing the mutational and copy-number landscape of the disease and identifying genetic correlates of 10-year local recurrence and radiotherapy response. This work is part of a large multi-institutional DCIS cohort programme conducted in collaboration with Sunnybrook Health Sciences Centre (Toronto).
A complementary methodological strand, developed jointly with Michael Hallett (Western), tackles the technical bottleneck that makes this work hard: FFPE-derived RNA-seq is sparse, degraded, and prone to extreme transcript dropout. We introduced generative and integrative modelling approaches that fit probabilistic distributions to FFPE expression counts, exploit matched-tissue profiles from the same patient, and use graph-attention over sample-sample adjacency networks to stabilize and impute missing values. These methods support ongoing development of RNA-based classifiers for invasive local recurrence and RT response on archival material that was previously considered too degraded to use.
Completed projects
Blood transcriptomics as a window onto exposure and disease
We have a long-standing interest in the development of RNA-based biomarker signatures in blood, which can be used to inform on exposure and health status. As a major defense and transport system, blood cells can adjust expression of their genes in response to various clinical, biochemical, and pathological conditions. To develop such surrogate signatures, we first defined robust laboratory methodologies for RNA profiling of blood cells (Dumeaux et al., 2008 Biomarkers in Medicine), and investigated normal inter-individual variation in healthy individuals (Dumeaux et al., 2010 PLoS Genetics). These studies served as stepping stones to several projects investigating how the signatures can be used to detect systemic molecular processes deregulated in response to defined exposure and health status (>5 publications, 200+ citations). These manuscripts highlight specific behavioral programs, such as metabolism or signaling, that are deregulated in an individual’s blood cells as biological or pathological responses to a given condition in the general population (e.g., exposure to organic pollutants) or in a diseased population (e.g., radiation-induced fibrosis in breast cancer survivors).
In earlier work, we showed that blood-based signatures could be used to develop new ways to diagnose disease. One line of that work produced a whole-blood diagnostic test for breast cancer (Dumeaux et al., 2015 IJC, plus one patent), and it helped motivate our current interest in single-cell approaches to blood-based biomarkers.
Selected papers
- Mutational landscape of pure ductal carcinoma in situ and associations with disease prognosis and response to radiotherapy
- Generative and integrative modeling for transcriptomics with formalin-fixed paraffin-embedded material
- Interactions between the tumor and the blood systemic response of breast cancer patients
- The prognostic ease and difficulty of invasive breast carcinoma
- Peripheral blood cells inform on the presence of breast cancer: a population-based case-control study
Selected software
- MIxT — system for exploring and comparing transcriptional profiles from two or more matched tissues across individuals.
- PREFFECT — generative and integrative modeling for FFPE transcriptomics.