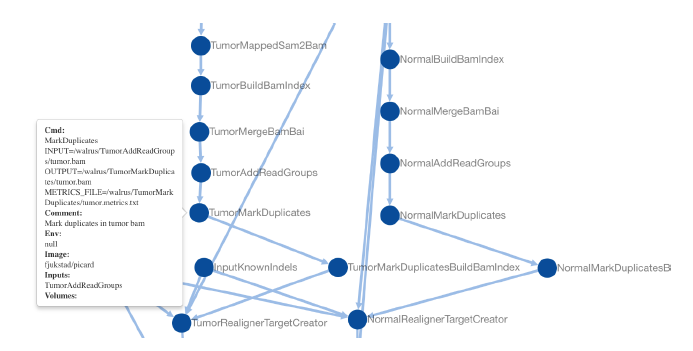
Precision medicine brings the promise of more precise diagnosis and individual-zed therapeutic strategies from analyzing a cancer’s genomic signature. Technologies such as high-throughput sequencing enable cheaper data collection at higher speed, but rely on modern data analysis platforms to extract knowledge from these high dimensional datasets. Since this is a rapidly advancing field, new diagnoses and therapies often require tailoring of the analysis. These pipelines are therefore developed iteratively, continuously modifying analysis parameters before arriving at the final results. To enable reproducible results it is important to record all these modifications and decisions made during the analysis process. We built a system, walrus, to support reproducible analyses for iteratively developed analysis pipelines. The approach is based on our experiences developing and using deep analysis pipelines to provide insights and recommendations for treatment in an actual breast cancer case. We designed walrus for the single servers or small compute clusters typically available for novel treatments in the clinical setting. walrus leverages software containers to provide reproducible execution environments, and integrates with modern version control systems to capture provenance of data and pipeline parameters. We have used walrus to analyze a patient’s primary tumor and adjacent normal tissue, including subsequent metastatic lesions. Although we have used walrus for specialized analyses of whole-exome sequencing datasets, it is a general data analysis tool that can be applied in a variety of scientific disciplines. We have open sourced walrus along with example data analysis pipelines at github.com/uit-bdps/walrus.