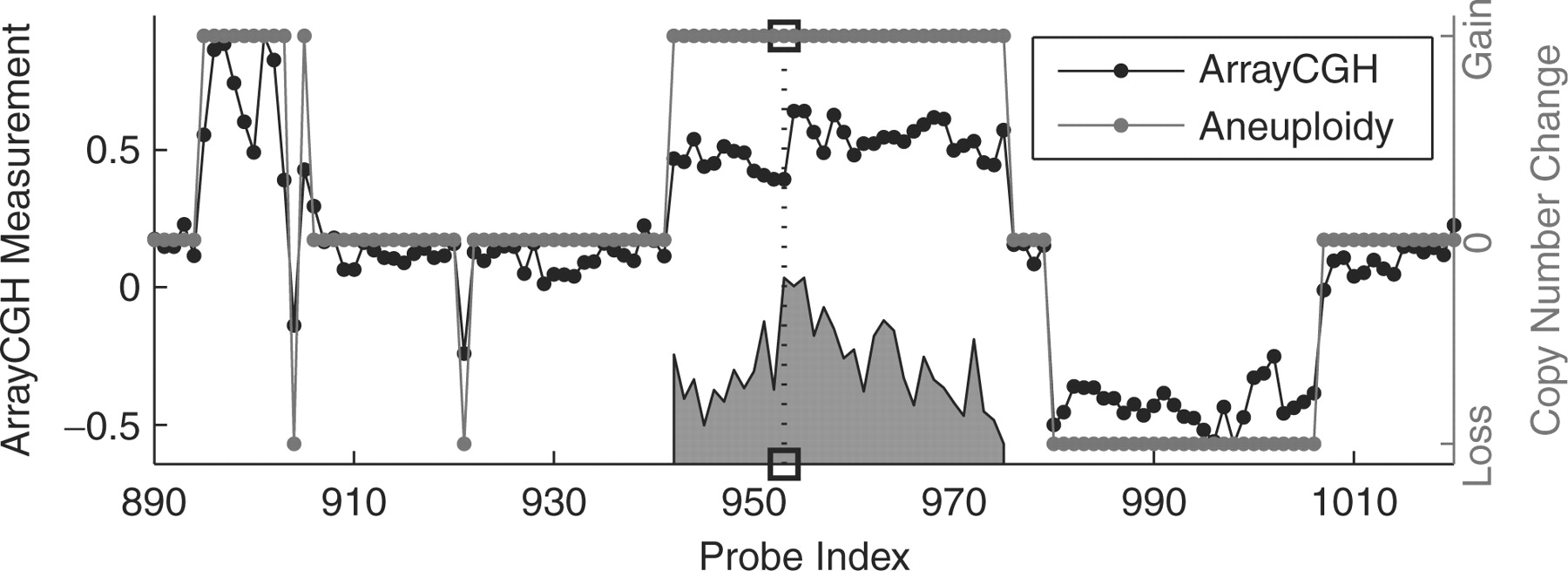
The heterogeneity of cancer cannot always be recognized by tumor morphology, but may be reflected by the underlying genetic aberrations. Array comparative genome hybridization (array-CGH) methods provide high-throughput data on genetic copy numbers, but determining the clinically relevant copy number changes remains a challenge. Conventional classification methods for linking recurrent alterations to clinical outcome ignore sequential correlations in selecting relevant features. Conversely, existing sequence classification methods can only model overall copy number instability, without regard to any particular position in the genome.
Here, we present the heterogeneous hidden conditional random field, a new integrated array-CGH analysis method for jointly classifying tumors, inferring copy numbers and identifying clinically relevant positions in recurrent alteration regions. By capturing the sequentiality as well as the locality of changes, our integrated model provides better noise reduction, and achieves more relevant gene retrieval and more accurate classification than existing methods. We provide an efficient L1-regularized discriminative training algorithm, which notably selects a small set of candidate genes most likely to be clinically relevant and driving the recurrent amplicons of importance. Our method thus provides unbiased starting points in deciding which genomic regions and which genes in particular to pursue for further examination. Our experiments on synthetic data and real genomic cancer prediction data show that our method is superior, both in prediction accuracy and relevant feature discovery, to existing methods. We also demonstrate that it can be used to generate novel biological hypotheses for breast cancer.